Paper: Whisper: read sorting allows robust mapping of DNA sequencing data
Summary
Short read alignment by sorting reads and using suffix array. Whisper does not follow the seed-and-extend paradigm. Sorting short reads requires initial cost, but pays on overall.
Contributions.
- Novel alignment algorithm based on Dirichlet principle and extensive use of sorting.
- High alignment accuracy, especially for indel (comparable to BWA-MEM or Minimap2)
Notes.
- Interesting idea with outstanding alignment speed
- Sorting reads does not take much time than expected.
- Given large memory (affects OS disk I/O caching) and high disk I/O, Whisper2 is much faster than reported (Deorowicz et al., SoftwareX, 2021).
- Searches upto K Leveneshtein errors given K+2 major stages (major stages have minor stages i, 0$\le$i$\le$ K).
Algorithms of Whisper
Major steps:
- Preprocessing reads
- Main processing
- Post processing
Step 1. Preprocessing
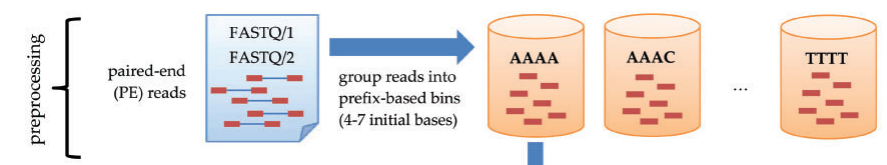
- Sort reads and store all reads in bins.
- Bins are divided by the prefix of the reads. Prefixes are established in a way that the distribution of the number of occurrences is uniform.
- Bins are generated by 4^maximum_prefix_length, then reduced until the total number of bins are less than input bin number threshold. Use priority queue to sort the total element number inside bins.
Step 2. Main Processing
Key insight is that, for read to align with at most K Levenshtein distance, there should be at least one substring (given short read divided into K+1 substrings) that exact matches to the reference.
- Cache-efficient because, reads and suffix array are both sorted.
- Bins are processed one-by-one, and part of suffix array corresponding to the bin is loaded partially.
- All mapping positions found is used in post processing step.
'''
@param1 sr: ShortRead
@param2 K: at most K Levenshtein errors
'''
Major_stage = K+2
Minor_stage = 0
shortread_length = len(sr)
for stage in Major_stage:
# Minor stages
for m_stage in stage:
# by Dirichlet principle there must exist one substring that exact matches to reference, if short read have at most K Levenshtein errors
# find exact match using suffix array and substring of short read
Find_exactmatch_in_suffixarray(sr[shortread_length*m_stage/stage: shortread_length*(m_stage+1)/stage])
Step 3. Post Processing
There are 4 steps inside post processing step.
- high-quality close mappings are filtered, appended to final alignment result.
- If step 1 fails, use Myers’ bit-parallel algorithm for Levenshtein distance.
- If step 1,2 fail, hash 20-mers of query read, match against reference.
- Pair mappings of each paired short read based on the Blast-like alignment scoring used in BWA-MEM.